
2018
Developing light-activated functional molecules has been a major goal in molecular engineering, as such systems hold the promise for solutions to many technological problems ranging from IT to sustainable development. Relevant efforts have been focused on engineering complexes of the abundant and environment-friendly iron, particularly on the ubiquitous model systems with polypyridine ligands. The first steps for developing efficient light-activated functional molecules is to obtain a thorough understanding of their potential energy surfaces (PES), and unveil the relaxation pathways on them after the excitation. We report on some theoretical and experimental results on iron based molecules which shall assist to open new paths for developing molecular systems which are rather promising for applications .
Quantum nuclear wavepacket simulations of photoexcited Fe-carbene complexes
Determining the possible photorelaxation pathways and estimating the lifetimes of the excited states is necessary for revealing the mechanism in functional molecules. A theoretical approach to this requires the simulation of the photorelaxation process, whereas for systems with a high density of excited states, such as several transition metal complexes, it might be necessary to explicitly account for the excitation process as well. Continuing our previous studies of iron complexes suggested as substitutes for the expensive and environmentally unfriendly rare earth-based photosensitizers for light-harvesting systems [Pápai et.al. J. Phys. Chem. Lett. 7 (2016) 2009, J. Phys. Chem.C 120 (2016) 17234] we report taking this aspect into account in the ultrafast excited state relaxation dynamics of the [Fe(btbip)2]2+ and [Fe(bmip)2]2+ (Figure 1) complexes.
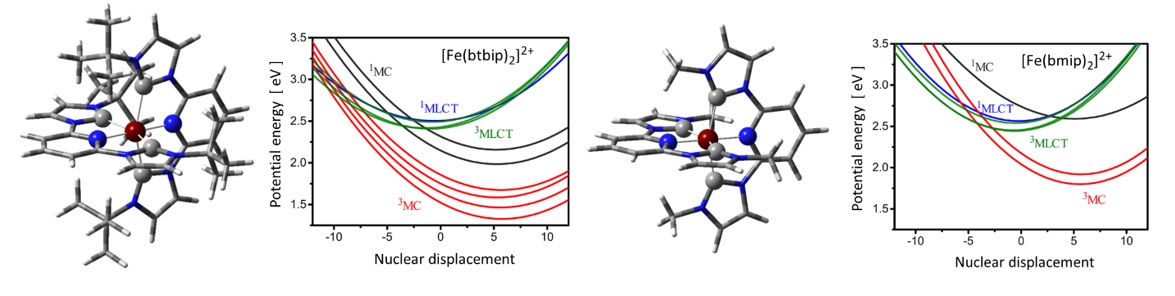
Figure 1. Molecular structures and potential energy curves for some excited states along the lowest frequency breathing vibrational mode of [Fe(btbip)2]2+ (btbip = 2,6-bis(3-tert-butyl-imidazole-1-ylidene)pyridine) (left) and [Fe(bmip)2]2+ (bmip = 2,6-bis(3-methyl-imidazole-1-ylidene) pyridine) (right). (N and C atoms are indicated by blue and grey colors, respectively.)
We performed quantum dynamics simulations in a four dimensional space spanned by the vibrational normal modes most relevant to the transitions. The current, improved description explicitly includes the interaction with the pump laser field in the Hamiltonian of the system in the semiclassical dipole approximation using transition dipole moments (TDM) obtained by the same time-dependent density functional theory (TD-DFT) method (B3LYP*/TZVP) used also for computing the potential energy surfaces and the spin-orbit couplings (SOCs). The simulations were performed with the multi-configurational time-dependent Hartree (MCTDH) method in the vibrational coupling Hamiltonian (VCHAM) formalism. In this formalism the SOCs and TDM vectors are directly determined by TD-DFT, while the non-adiabatic coupling terms and the parameters of the multidimensional potential energy surfaces are determined by fitting to TD-DFT adiabatic potential energy values. In both molecules, the optically bright singlet metal-to-ligand charge transfer state (1MLCT), as well as the energetically closest singlet metal-centred (1MC) states are degenerate, showing E symmetry. We have demonstrated that the spin-vibronic model, constructed directly from electronic structure calculations as described above, can exhibit erroneous, polarization-dependent relaxation dynamics for such degenerate states due to artificial interference of coupled relaxation pathways. We have shown that this problem stems from an incorrect description of the excitation of the ground state nuclear wavepacket into electronically degenerate states. The reason for this deficiency is that the simulation of the excitation process lacks rotational invariance implied by the symmetry of degenerate states. This translates into unphysical behavior through the nonadiabatic couplings among these states. We have demonstrated that a proper complex representation of TDMs is necessary to ensure rotational invariance of the excitation. This eliminates the unphysical interferences and thus produces correctly polarization-independent excited-state dynamics for both investigated complexes. Figure 2 shows the population dynamics for four simulations with different laser polarizations for which - without the correction - we obtained more than 20% deviations in excited state populations at 2 ps after the excitation and a difference of almost a factor of three in relaxation times.
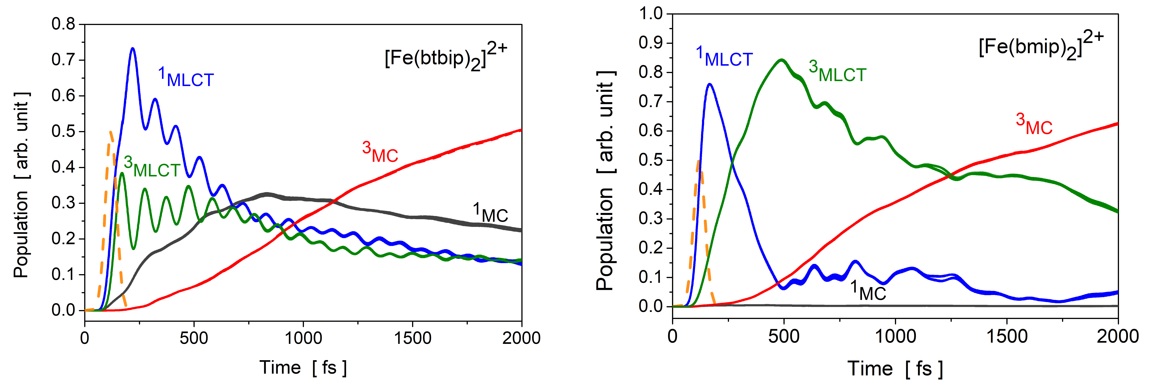
Figure 2. Results of the simulations: Excited state populations as a function of time for the [Fe(btbip)2]2+ (left) and [Fe(bmip)2]2+ (right) complexes following excitation by a 485 nm, 60 fs Gaussian pulse with four different laser polarizations. (The temporal intensity profile of the laser pulse is indicated by dashed orange line.) Due to the corrected description of the excitation process, no polarization dependence is observed. The paper can be downloaded from http://dx.doi.org/10.1021/acs.jctc.8b00135.
In agreement with related experiments [Liu et.al. Chem. Comm. 49 (2013) 6412] the simulations reveal that an apparently minor structural difference considerably alters the relaxation dynamics in these functional complexes. In the present case, the tert-butyl group stabilizes the 1MC states, enabling the 1,3MLCT → 1MC population transfer in addition to the 3MLCT → 3MC pathway, which in turn leads to much faster relaxation of MLCT states. This explains the different photophysical behavior of these otherwise similar complexes, pointing out a crucial issue for tailoring the function in such complexes.
Controlling the relaxation pathway in photoexcited transition metal complexes via substitution and solvent effects
An alternative way to alter the photophysical properties of transition metal complexes is to shift the potentials with solvent interactions. This can change the intersections of the potentials of and the couplings between different states, which will accordingly influence the transition trajectories and probabilities. Replacing two bipyridine ligands in the frequently studied [Fe(bpy)3]2+ (bpy = 2,2’-bipyridine) by 4 monodentate CN− ions introduces two effects that allows us to modify the PES. First, the CN− substitution makes the ligand field stronger, which shifts the MC (metal centered) states to higher energy; second, interaction with protic solvents affects the strength of the Fe−C bonds, which shifts the energy of the MLCT (metal-to-ligand charge transfer) states. We have investigated these effects on the [Fe(bpy)(CN)4]2− complex, in collaboration with the groups of Prof. Kelly Gaffney (SLAC) and Prof. M. Nielsen (DTU Lyngby), using a combination of transient optical absorption (TOAS) and X-ray emission spectroscopies. The first effect observed is that due to the increase in the ligand field, the MC states are destabilized, thus the potential energy landscape, as well as the relaxation pattern changes completely. While for [Fe(bipy)3]2+, the relaxation follows a pattern the for the full photocycle 1GS ® 1MLCT®3MLCT®3MC ®5MC ® 1GS, with very short lived excited singlet and triplet states (τ < 100 fs), and a long lived (600 fs) quintet, the field enhancement of the 4 CN ligands shifts up the quintet to an unreachable energy, and makes the triplets longer lived. However, it depends on the solvent which triplet is stabilized. In water, a solvent with strong Lewis acidity/H-bonding ability, the MLCT excited state of [Fe(bpy)(CN)4]2− decays still rapidly, in less than 100 femtoseconds, forming a quasi-stable metal-centered excited state with 13 picosecond lifetime, while in weak Lewis acid solvents, such as dimethyl sulfoxide (DMSO) or acetonitrile, the same molecule possesses 19 picosecond 3MLCT excited state lifetime and no discernible contribution from MC states. It was shown that the MC excited state in the former case has triplet (3MC) character, unlike other reported six-coordinate Fe(II)-centered coordination compounds with a singlet ground state forming MC quintet (5MC) states. The influence of the solvent on the excited state relaxation dynamics arises from different solvent stabilization of the CN− ligand,
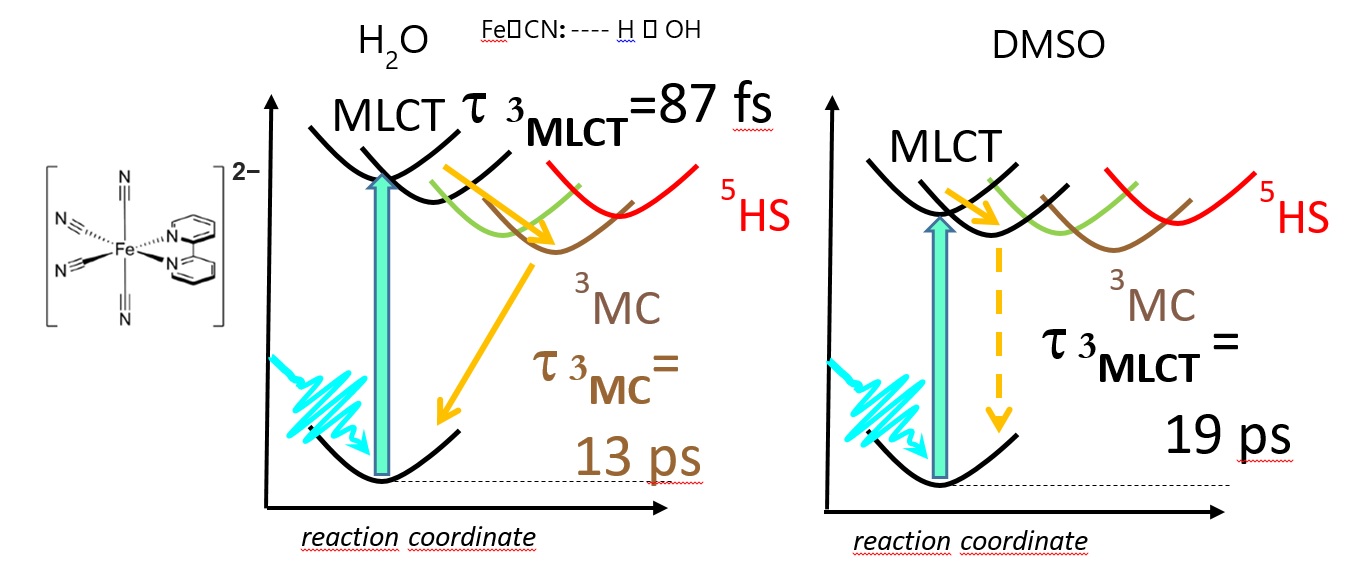
Figure 3. The [Fe(bpy)(CN)4]2− molecule (left), and its schematic potential energy curves for water (middle) and DMSO (right) solvents. Yellow arrows show the relaxation pathways. (The full article can be accessed at http://dx.doi.org/10.1039/c7cp07838b.)
which in turn modifies the relative energies of the MLCT potential energy curves, as shown in Figure 3. The observed solvent dependent changes in excited state non-radiative relaxation for [Fe(bpy)(CN)4]2− allows us to infer the influence of the solvent on the electronic structure of the complex.
Following the results detailed above, we have chosen to carry out a similar substitution on another model system, [Fe(terpy)2]2+ (terpy: 2,2':6',2''-terpyridine) complex, whose photophysical behavior is well known [Pápai, M. et al., J. Chem. Theory Comput. 9, 509. (2013); Vankó, G. et al., J. Phys. Chem. C, 119 5888. (2015)]. The effect of the terpy – CN ligand substitution should in principle be the similar as described above for the bipyridine analogue, thus it should allow us to verify this type of controlling of the PES, hence the elementary processes of photorelaxation. Our theoretical (TD-)DFT and transient optical absorption studies on the [Fe(terpy)(CN)3]− system confirmed relevant variations in the PES and the time evolution, respectively, see the upper part of Fig. 4. Upon the cyanide substitution in [Fe(terpy)2]2+, the calculated triplet and quintet MC states are shifted to higher energies and the solvent effect on the MLCT bands is also reproduced (not shown here). TOAS reflect significantly different behavior compared to the original [Fe(terpy)2]2+, and also some difference from the bipy/CN mixed complex discussed above. Here the ligand field enhancement is somewhat smaller because of the smaller number of CN ligands, thus the quintet MC state is still reached in the relaxation, yet its small yield reflects small branching into this state. During one of the first pilot experiments at the European X-ray Free Electron Laser it was possible to perform a quick feasibility test on aqueous [Fe(terpy)(CN)3]− with 515 nm laser excitation and collect a delay scan of difference signals on the Fe Kα lines. The lifetime obtained is fully consistent with that of the major component in the TOAS signal; however, better statistics, longer scan range and the collection of the Kβ spectrum and wide angle scattering are required to fully characterize the excited state dynamics in these molecular systems.
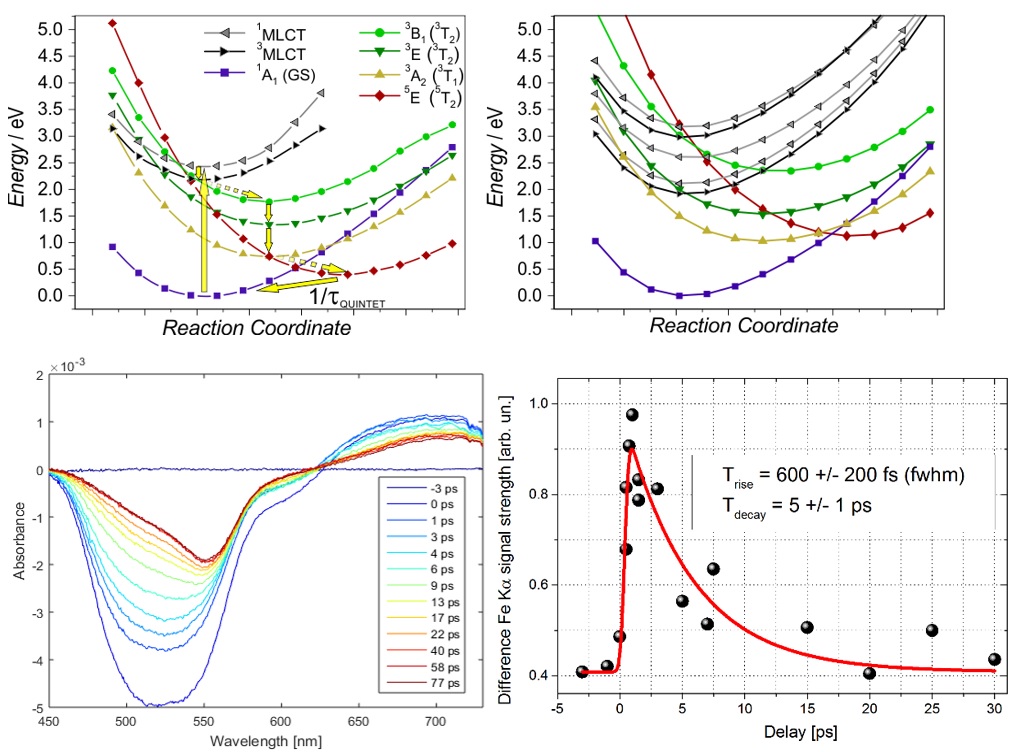
Figure 4. TOP: Calculated PES of the relevant ground-, MLCT, and MC excited states for [Fe(terpy)2]2+ (left) and for [Fe(terpy)(CN)3]−(right). BOTTOM: TOAS (left) and pilot XFEL data: Kα XES difference signal of [Fe(terpy)(CN)3]− in water as a function of time delay (right).
