2021
Developments — The group has been continuously developing theoretical and experimental tools in order to investigate the ultrafast photoinduced dynamics in functional molecules, and to design new ones with improved performance. The experimental tools include pioneering static high-energy-resolution laboratory X-ray spectroscopy, with hard X-ray absorption being performed routinely and efficiently, while X-ray emission spectroscopy is still being optimized. Ultrafast studies with X-ray spectroscopy and scattering probes are carried out at synchrotron radiation sources and X-ray free electron lasers, but we also participate in X-ray developments at the Extreme Light Infrastructure. In addition, we have realized in our home laboratory a femtosecond-resolved transient optical absorption spectroscopy station, and a femtosecond stimulated Raman scattering setup is being built. Our theoretical efforts focus on designing new functional molecules with quantum chemistry (which we later attempt to synthesize and then subject to experimental tests), as well as running (quantum) dynamics simulations to model the dynamics in light activated molecular systems. In what follows we report such works that were published in the previous year.
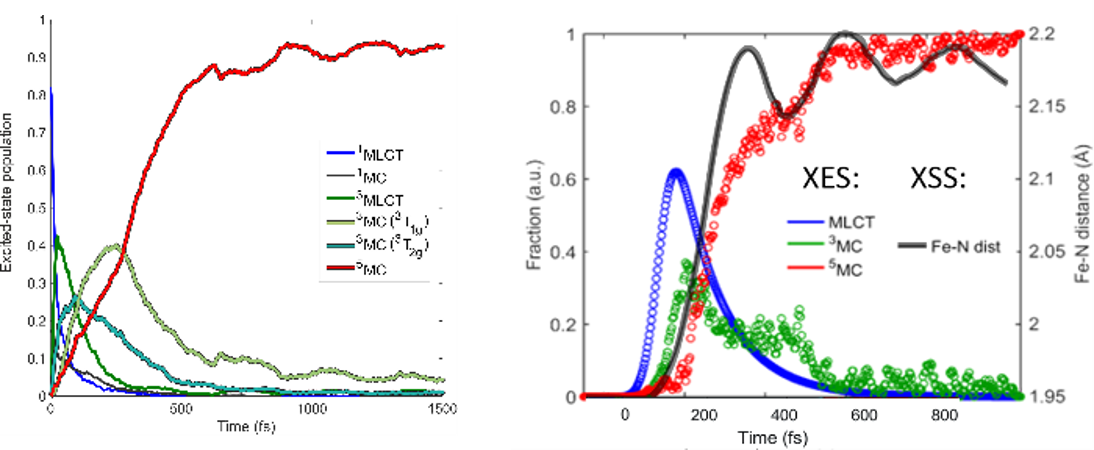
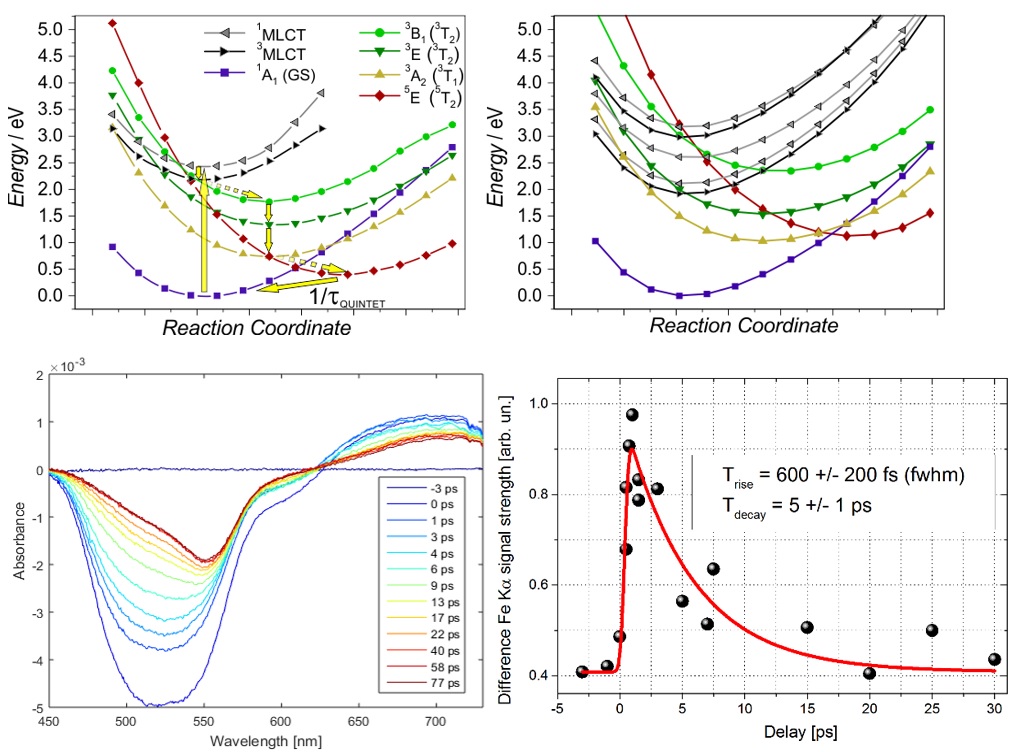
Simulation of ultrafast excited-state dynamics in molecules. — Time-resolved (TR) pump-probe experiments are very powerful tools to resolve ultrafast excited-state dynamics in functional molecules. However, the recorded TR data is very often complex and difficult to interpret, computations thus have a crucial role in extracting all important information from the measured data. Theory offers two fundamentally different approaches for the simulation of excited-state dynamics: i) quantum dynamics (QD), which is exact but only feasible for a few degrees of freedom, one is thus forced to develop reduced dimensionality models for molecules, and ii) semiclassical dynamics, which allows full dimensionality but in general, does not account properly for quantum effects. In an international collaboration, we employed trajectory surface hopping (TSH), which belongs to group ii), to simulate the photoinduced dynamics of trimethylamine (TMA) in full dimension, initiated from and occurring via Rydberg excited states [X1]. Importantly, the dynamics of TMA involve dissociation, for which the widely-utilized vibronic-coupling model of QD fails, but TSH does not. The utilized methodology is based on on-the-fly (time-dependent) density functional theory (DFT/TD-DFT) potential energy surfaces (PESs) and an energy-based technique to force an electronic transition near the S1/S0 conical intersection (CI), in order to avoid single-reference failure of DFT at the CI. We concluded that electronic excitation into Rydberg states induces strong coherent vibrational umbrella motion, whose decoherence dynamics, in turn, controls the electronic relaxation and ballistic dynamics along the N-C bond. These N-C stretching dynamics can potentially open up for photodissociaton, which is controlled by the branching at the S1/S0 CI; we also found that the simulated dissociation yield depends heavily on the applied exchange-correlation functional. In a second work, we presented the first ab initio simulation of the entire singlet-triplet-quintet dynamics utilizing a synergistic TSH-QD methodology [X2]. For this study, we chose the octahedral [Fe(NCH)6]2+ complex, which is a model for Fe(II) low-spin (LS) → high-spin (HS) photoswitching solely involving metal-centered excited states (Figure Xa). We here used full-dimensional TSH to select modes (see Figure Xb) for QD based on PESs and couplings calculated by high-level multiconfigurational quantum chemistry (CASPT2). The obtained mechanistic picture is consistent with experimental findings and includes assignment of the principal intermediate 3T2g, whose dynamics are controlled by the key Fe-N stretching vibrations.
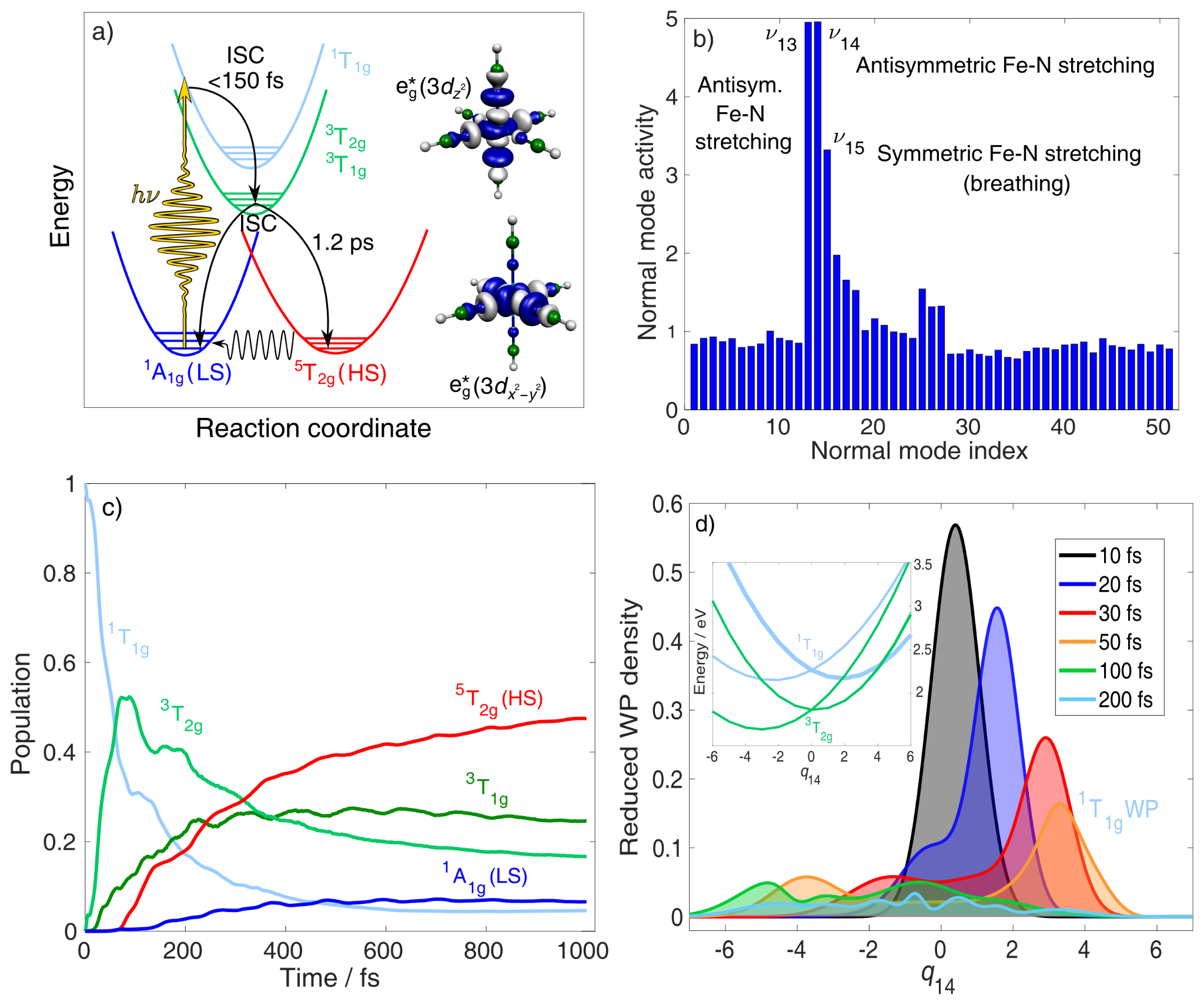
Figure X. Summary of the synergistic spin-vibronic simulations on the octahedral model [Fe(NCH)6]2+ for LS → HS photoswitching. (a) Schematic mechanistic picture with the antibonding eg* orbitals illustrating the metal-center character of the excited states. (b) Mode selection by full-dimensional TSH (TD-DFT). (c) Population dynamics by 3D QD (CASPT2). (d) QD (CASPT2) along the antisymmetric Fe-N stretching normal mode coordinate q14, illustrating singlet → triplet ISC and wavepacket spreading. Adapted from ref. X2, the full article can be accessed at: https://doi.org/10.1021/acs.inorgchem.1c01838.
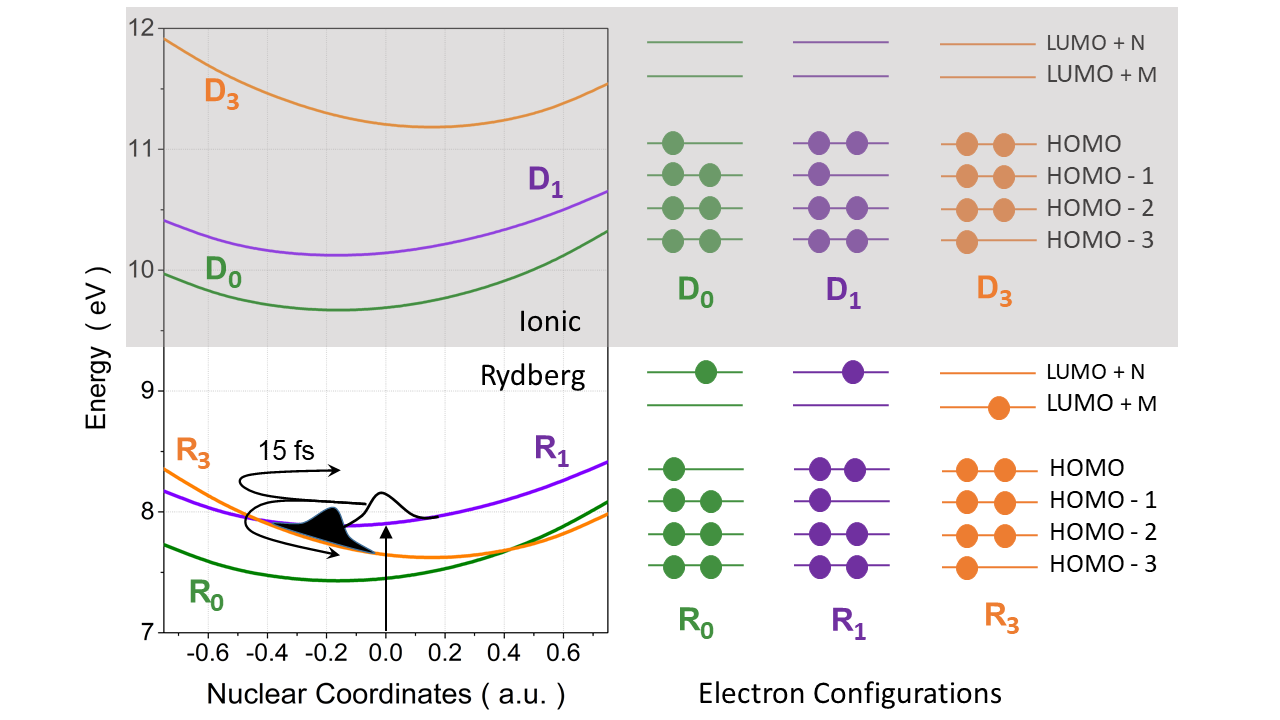
Fs-resolved experiments and theoretical interpretation of nonadiabatic dynamics for small benchmark molecules. — Calculation of time-dependent experimental observables, based on excited-state dynamics simulations, can be extensively used to interpret TR data. X-ray absorption spectroscopy (XAS) is an element and chemical state specific technique that is able to resolve electronic states and transitions between them. In an international collaboration, we assessed the performance of different electronic structure methods for the calculation of excited-state XAS of three benchmark molecules: uracil, thymine, and acetylacetone [X3]. We found that TD-DFT (B3LYP), utilizing the maximum overlap method (MOM) to obtain core-excited configurations that are inaccessible for TD-DFT referenced to the ground state, performs reasonably well. In combination with TSH, we used MOM-TD-DFT to simulate the TR-XAS of pyrazine, promoted to the 1B2u (ππ*) to interpret a fs-resolved experiment. We found that in addition to 1B2u (ππ*) and 1B3u (nπ*) states, the debated optically-dark 1Au (nπ*) state also participates in the electronic relaxation of pyrazine. We identified experimentally that the 1Au state is populated in 200±50 fs and is assigned by the 284.5 eV shoulder, red-shifted from the ground-state main peak by 0.9 eV, which, based on the XAS calculations, can only be attributed to 1Au state (Figure Yc).
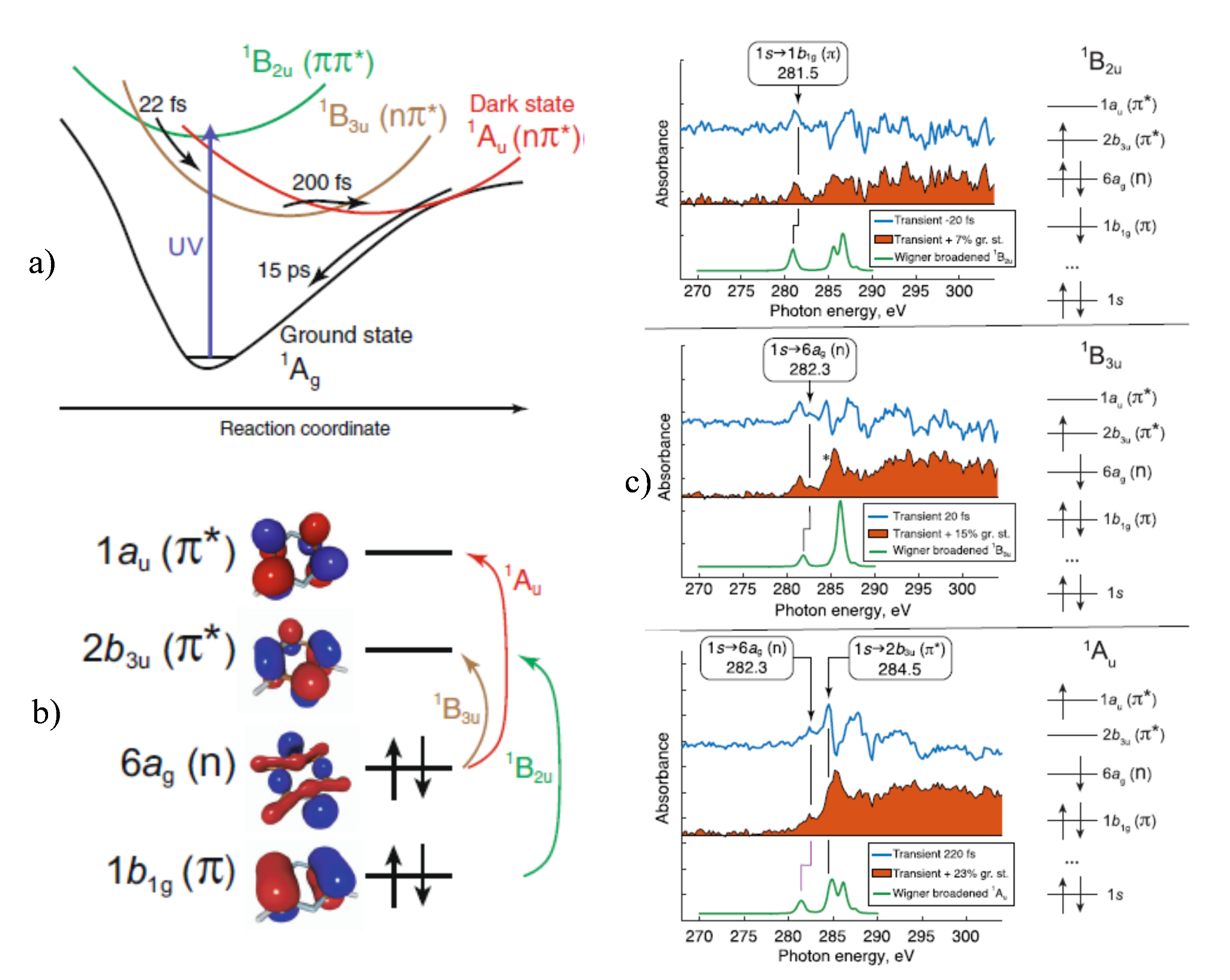
Figure Y. a) Schematic illustration of the excited-state dynamics of pyrazine with the experimental timescales; b) Dominant electronic character of the valence states 1B2u, 1B3u, and 1Au; c) Experimental difference XAS (blue, pump-on − pump-off ), the excited-state spectra, obtained by correction for ground-state bleach (orange filled area), and the simulated XAS for three selected time delays −20 fs, 20 fs, and 220 fs.
Also in an international cooperation we investigated the effect of non-adiabatic vibrational dynamics on resonantly enhanced multiphoton ionization. Time-resolved photoelectron spectroscopy (TR-PES) measurements on the benchmark CH2BrI molecule were interpreted in terms of 1D quantum dynamics simulation of the strong-field ionization dynamics, which include multiphoton resonance, dynamic Stark shifts, as well as vibrational dynamics and internal conversion during the ionization process. The theoretical model was based on the combination of the adiabatic elimination of off-resonant electronic states with the discretization of the ionic continua by Legendre polynomials of the photoelectron's kinetic energy. In agreement with the measured photoelectron spectra the simulations demonstrated how the ionization process can be controlled by phase-modulation of the exciting femtosecond near-infrared pulses. [Y1]
References:
[X1] Mátyás Pápai, Xusong Li, Martin M. Nielsen, Klaus B. Møller Trajectory surface-hopping photoinduced dynamics from Rydberg states of trimethylamine Phys. Chem. Chem. Phys., 23 (2021) 10964−10977.
[X2] Mátyás PápaiPhotoinduced Low-Spin High-Spin Mechanism of an Octahedral Fe(II) Complex Revealed by Synergistic Spin-Vibronic DynamicsInorg. Chem. 60 (2021) 13950−13954.
[X3] Shota Tsuru, Marta L. Vidal, Mátyás Pápai, Anna I. Krylov, Klaus B. Møller, Sonia Coriani An Assessment of Different Electronic Structure Approaches for Modeling Time-Resolved X-ray Absorption Spectroscopy Struct. Dyn. 8 (2021) 024101, (Special Issue „Theory of Ultrafast X-ray and Electron Phenomena”)
[X4] Valeriu Scutelnic, Shota Tsuru, Mátyás Pápai, Zheyue Yang, Michael Ephstein, Tian Xue, Eric Haugen, Yuki Kobayashi, Anna I. Krylov, Klaus B. Møller, Sonia Coriani, Stephen R. Leone X-ray transient absorption reveals the 1Au (nπ*) state of pyrazine in electronic relaxationNat. Commun. 12 (2021) 5003.
[Y1] Brian Kaufman, Tamás Rozgonyi, Philipp Marquetand, Thomas Weinacht Competition between dynamic resonance and internal conversion in strong-field molecular ionization with chirped ultrafast laser pulsesPhys. Rev. A 103 (2021) 023108.